简介
BSA-Seq(Bulked Segregant Analysis Sequencing)工作流程是一种用于从F2群体的重测序数据中识别因果突变的遗传学分析方法。这种方法特别适用于植物或其他自交物种的QTL(数量性状基因座)定位和突变鉴定。
主要流程
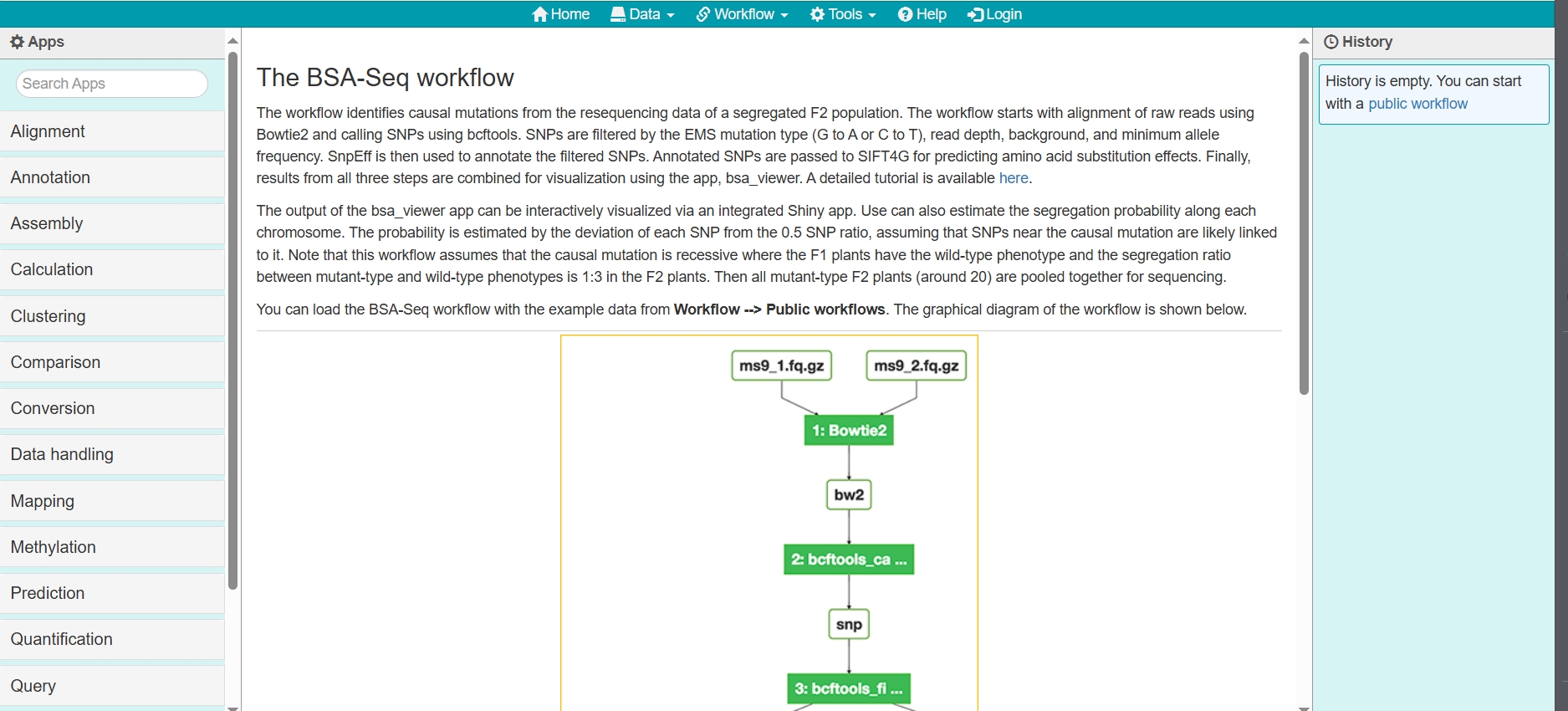
原始读取对齐:使用Bowtie2对原始读取序列进行对齐,确保它们与参考基因组匹配。
SNP调用:使用bcftools进行SNP(单核苷酸多态性)的识别和调用。
SNP过滤:根据EMS(乙基甲磺酸)突变类型(如G到A或C到T)、读取深度、背景和最小等位基因频率等标准对SNP进行过滤。
SNP注释:使用SnpEff对过滤后的SNP进行功能注释,以了解这些变异可能对基因和蛋白质的影响。
预测氨基酸取代效应:将带注释的SNP传递给SIFT4G,预测它们引起的氨基酸取代对蛋白质功能的影响。
结果整合:将上述步骤的结果结合起来,为后续的可视化和分析做准备。
可视化:使用bsa_viewer应用程序对结果进行可视化,通过集成的Shiny应用实现交互式可视化。
分离概率估算:估算每条染色体的分离概率,基于每个SNP与0.5 SNP比率的偏差,假设因果突变附近的SNP很可能与之相关。
隐性突变假设:工作流程假设因果突变为隐性突变,F1植株具有野生型表型,F2植株中突变型和野生型表型的分离比为1:3。
测序:将所有突变型F2植物(约20株)集中在一起进行测序。
使用场景
植物遗传学研究:适用于植物遗传学家在QTL定位和突变鉴定中使用。
自交物种研究:适用于自交物种的遗传分析,如拟南芥等模式植物。
结论
BSA-Seq为植物遗传学家提供了一种高效的方法来识别和分析F2群体中的因果突变。通过一系列的生物信息学工具和步骤,研究人员可以更准确地定位基因座并预测突变的生物学效应。该工作流程的自动化和可视化特点,使得即使是没有深厚生物信息学背景的研究人员也能够轻松使用。通过这种方法,研究人员可以更深入地理解遗传变异对植物性状的影响,推动植物遗传学和育种研究的发展。